
这个案例是讲药品GMP无菌制剂车间改造的
摘要:以一个无菌制剂车间的改造为例,对新版药品GMP中无菌药品附录的生产要求进行讨论,从工艺设计的角度分析,使改造设计满足新规范要求。
关键词:新版药品GMP;无菌制剂车间;设计
《药品生产质量管理规范(2010年修订)》已于2010年10月19日经卫生部部务会议审议通过,并与2011年3月1日起施行。本次修订涉及基本要求以及无菌药品、中药制剂、原料药、生物制品和血液制品五个附录,其中无菌药品是本次规范修订的一个重点,变化也最大。随着新版药品GMP的施行,大批无菌药品生产车间面临改造,如何使我们的设计符合新规范的要求,从硬件上保证无菌药品生产的安全及工艺流畅,对于设计人员是一个新的课题。本文以实际设计中的一个无菌制药车间的改造为例,在工艺方面对新版药品GMP的无菌药品生产要求进行分析和探讨。
1、新版药品GMP无菌药品生产相关要点
无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料包,包括无菌制剂和无菌原料药,根据生产工艺又可分为最终灭菌产品及非最终灭菌产品两类。为保证无菌药品的安全和质量提供法规和科学依据,新版规范采用了欧盟和WHO的A、B、C、D分级标准,对无菌药品生产的洁净度级别提出了非常具体的要求悬浮粒子的静态、动态监测、浮游菌、沉降菌和表面微生物的监测,细化了培养基模拟灌装、灭菌验证和管理的要求,增加了无菌操作的具体要求,强化了无菌保证的措施。
较原98版GMP标准,无菌药品生产环境的洁净度要求提高了。在洁净区分级上,原有的100级、1万级(包括无菌1万级)、10万级、30万级等,均改为与欧盟标准相当的A、B、C、D级。
A级是指高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或链接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。B级指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。C级和D级指无菌药品生产过程中重要程度较低操作步骤的洁净区。洁净区的洁净度要求分“静态”和“动态”标准,同时对洁净区的悬浮粒子和微生物进行动态监测。各级别空气悬浮粒子的标准规定如表1
表1 各级洁净度空气悬浮粒子标准
| 洁净度级别 | 悬浮粒子最大允许数/ m³ | |||
| 静态 | 动态 | |||
| ≥0.5μm | ≥5.0μm | ≥0.5μm | ≥5.0μm | |
| A级 | 3520 | 20 | 3520 | 20 |
| B级 | 3520 | 29 | 352000 | 2900 |
| C级 | 352000 | 2900 | 3520000 | 29000 |
| D级 | 3520000 | 29000 | 不作规定 | 不作规定 |
2、无菌制剂车间改造实例
广东某制药有限公司一无菌制剂车间,生产非最终灭菌产品,现须根据新版GMP的要求,对原车间进行原地的改造设计。原车间的设备平面布置图间图1。
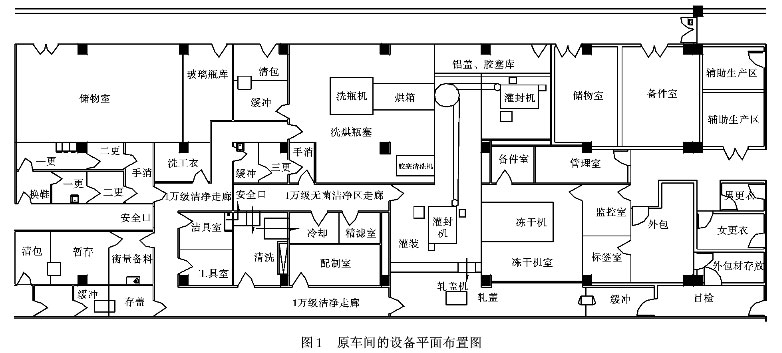
对非最终灭菌的产品,由于产品装入最终容器后,不再作进一步的灭菌处理,因此必须对整个无菌药品的生产过程及其环境条件进行严格的控制,以保证整个生产过程不被微生物所污染。具体操作分级见表2。
表2操作分级表
| 洁净度级别 | 非最终灭菌产品的无菌生产操作示例 |
| B级背景下的A级 |
1.处于未完全密封状态下产品的操作和转运,如产品灌装(或灌封)、分装、压塞、轧盖等; 2.灌装前无法除菌过滤的药液或产品的配制; 3.直接接触药品的包装材料、器皿灭菌后的装配以及处于未完全密封状态下的转运和存放; 4.无菌原料药的粉碎、过筛、混合、分装。 |
| B级 |
1.处于未完全密封(1)状态下的产品置于完全密封容器内的转运; 2.直接接触药品的包装材料、器皿灭菌后处于密闭容器内的转运和存放。 |
| C级 |
1.灌装前可除菌过滤的药液或产品的配制; 2.产品的过滤 |
| D级 | 直接接触药品的包装材料、器具的最终清洗、装配或包装、灭菌。 |
其中轧盖前产品视为处于未完全密封状态。根据已压塞产品的密封性、轧盖设备的设计、铝盖的特性等因素,轧盖操作可选择在C级或D级背景下的A级送风环境中进行。A级送风环境应当至少符合A级区的静态要求。
2.2原车间改造要点
原车间在工艺布局上有几个不足之处,也不能满足新版规范要求,以下作分析讨论。
(1)洗瓶灭菌区不应与配液在同一级别区域进行生产,灌装前可除菌过滤的药液或产品的配制工序,相对洗瓶灭菌的生产过程及其环境条件要严格,洁净度要求也相应要高,如果在同一区域生产,生产人员穿插于2个不同要求的工序,共用同一人员净化通道,生产中使用的容器具共用同一清洁间清洗,容易对配液工序产生影响,在工艺和空调上会容易造成污染。按照新GMP要求,配液须设置在C级;洗瓶灭菌应该设置在D级区。
(2)无菌生产核心区需要考虑A级保护,包括了无菌药品的灌装区、灭菌后的瓶、塞、盖进入无菌操作的区域、灌装后半加塞产品等待冻干及冻干后产品暂存区域、无菌过滤器的连接组装区域等;进入无菌区的工器具清洗后等待灭菌区,以及灭菌后的存放区域,如果没有采取其他密封保护措施,直接暴露的,也需加设A级送风保护。
(3)由于轧盖前产品视为处于未完全密封状态,因此操作需要在A级的环境保护下进行,轧盖区同样属于无菌生产核心区。但是由于轧盖会产生大量非活性的微粒,会随着气流飞扬,粘在设备和操作人员人体表面,在生产环境保护的同时也是一个污染源,若与灌装操作区放在同一区域,使用同一B级区的空调系统,则会造成产品和生产环境的污染。因此在布局上,考虑轧盖操作区独立设置,人员单独更衣,在气流组织上,轧盖区相对灌装区负压;考虑到该工序的特殊性,可设置为C级背景下的A级送风保护下进行操作。
综合以上几点,该无菌产品的生产流程及洁净分区示意图见图2。
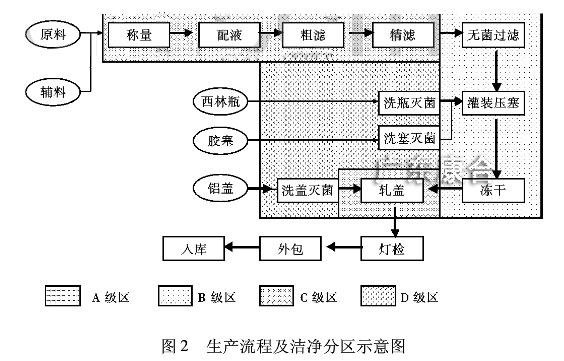
(5)加设B、C、D级各区废品通道,B级区废弃物由缓冲室传递柜传出;考虑到生产中人工操作需使用到哦周转盘,加设一般区的周转盘清洗间,再进入C级区精洗、灭菌。对应B级区的清洁消毒,在C级区设置消毒液配制间,过滤后进入B级区精滤后使用。
2.3改造后无菌制剂车间布局
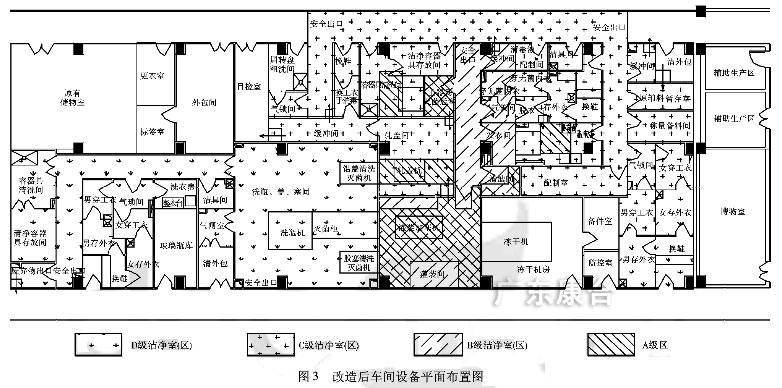
考虑到设备承重基础及原有配套公用工程管道的要求,尽量减少重大改动,因此不改变冻干机原有的摆放位置。按《建筑设计防火规范》的要求,加设洁净区的消防安全出口。据《建筑设计防火规范》的要求,加设洁净区的消防安全出口。据上述几个设计要点对原车间改造设计,改造后的无菌制剂车间设备平面布置图见图3。
新版GMP规范作为质量管理体系的一部分,是药品生产管理和质量控制的基本要求,旨在最大限度地降低药品生产过程中污染、交叉污染以及混淆、差错等风险,确保持续稳定地生产出符合预定用途和注册要求的药品。一个新规范的施行,必然带来新的技术和新的观念,对于设计人员须在实践中不断地进行探索与修正。本文仅从无菌制剂方面去进行分析,对新版药品GMP中无菌药品附录的工艺生产要求进行讨论,使设计能够满足新规范的要求。广州化工2011年39卷第14期,作者:李颖君
无菌制剂车间改造; 药品GMP无菌车间



